All published articles of this journal are available on ScienceDirect.

A Narrative Review of Exercise-Induced Oxidative Stress: Oxidative DNA Damage Underlined
Abstract
This is a narrative review principally aimed to highlight the factors related to exercise that induce oxidative DNA damage. Contracting skeletal muscles during exercise involve increased production of reactive oxygen species (ROS) from different cellular sources. Exercise-induced oxidative stress causes damage to cellular macromolecules proteins, lipids, and DNA. To date, oxidative DNA damage has been minimally investigated. The exercise-induced oxidative DNA damage topic was introduced first by discussing ROS chemistry, sources, and cellular damaging effects. It appears that exercise modality, intensity, duration, and the conditions under which the exercise was performed play major roles in determining the level of oxidative DNA damage during exercise. This review also discusses the possible mechanisms by which regular exercise protects against oxidative DNA damage.
1. INTRODUCTION
Regular physical activity is well-known for reducing the risk of cardiovascular diseases and several types of cancer [1]. In contrast, reactive oxygen species (ROS) accumulated during exhaustive exercise are associated with increased risk of numerous pathologies such as carcinogenesis, diabetes, and cardiomyopathy [2]. Exercise-induced oxidative stress is a term indicating an imbalance between oxidants and antioxidants [3]. Aerobic exercise of different intensities from as short as 5 min to ultra-endurance races has been shown to elicit wide ranges of ROS production [4, 5], with evidence suggesting the higher intensity as a determining factor in exercise-induced ROS production [6]. Likewise, resistance exercise has been shown to generate ROS. Nonetheless, resistance exercise induces ROS production when performed at > 60% of the one-repetition maximum (1RM) [7, 8], whereas high repetitions (70 – 80 repetitions) at lower intensities induced a minimal, if any, rise in ROS production [9].
Exercise-induced ROS production can readily attack all cellular macromolecules, i.e., proteins, lipids, and DNA, resulting in a temporary decrease in muscular function. Paradoxically, low-to-moderate levels of ROS during exercise are involved in cellular signalling and regulatory roles in force production and several adaptations prompted with regular exposure to exercise [10]. While most studies have highlighted the damaging effect of exercise-induced oxidative stress on amino acids and lipids, nucleic acids have received less attention. Therefore, oxidative DNA damage during exercise is under the spotlight of this review. The flowchart of the review process is illustrated in Fig. (1). The articles investigating the effect of exercise on oxidative DNA damage were selected if they were published in a peer-reviewed journal, conducted in humans, used aerobic and/or resistance exercise modalities, and used 8-hydroxydeoxyguanosine and/or comet assay to assess the oxidative DNA damage.
2. CHEMISTRY OF FREE RADICALS
A free radical is a molecule or a single atom with unpaired electrons in the outermost shell. Free radicals are not capable of independent existence, are highly reactive, and are short-lived [11]. They can be formed by electron transfer, as well as by homolytic and heterolytic fission [12]. Homolytic bond cleavage typically requires energy in the form of UV light or heat. Alkyl and acyl peroxides, compounds containing –O—O– single bonds, can work as radical initiators because of their distinct low homolytic bond dissociation energy. For instance, hydrogen peroxide H2O2 (HO—OH) homolytic bond dissociation requires 214 kJ/mol compared to H2O (H—O—H), which requires 499 kJ/mol, suggesting the high susceptibility of –O—O– single bonds to form oxygen molecule radicals [11].
The unpaired electron of the free radical tends to attain the most favourable energetic state by binding with another electron of opposite spin. This reaction depends on the reaction-specific rate constant and the concentration of the reacting species. The free radical reacts with another radical or molecule, forming a radical-radical or radical-molecule product. The free radical has a high tendency to attack other cellular macromolecules in the vicinity of its production site [13].
2.1. Major Reactive Species and their Sources of Production during Exercise
Oxygen and nitrogen reactive derivatives are modestly produced during rest and intensely during vigorous exercise in skeletal and cardiac muscles [10]. Superoxide anion (O2•–) and nitric oxide (NO) are the main initiating free radicals that react with other molecules to produce further reactive oxygen and nitrogen species [14, 15]. Complex I of the electron transport chain is the main site of O2•– formation through electron transfer to O2 [13, 14]. Although O2•– is generally impermeable to the cell membrane, its relatively long half-life and susceptibility to protonation at physiological pH increase its likelihood to transfer across the cellular membrane [10, 16]. However, it has been argued whether superoxide protonation can ever occur under pH 7.4 conditions [17]. Superoxide is a potential reducing and oxidizing agent: it can reduce ferric complexes such as cytochrome c [18] and oxidize other molecules such as ascorbic acid [19]. Conversely, in aqueous solutions, it is a weak oxidizing agent and rather a stronger reducing agent [16]. Superoxide can readily undergo catalyzed and non-catalyzed dismutation to produce a relatively stable long-half life and membrane-permeable hydrogen peroxide (H2O2) reactive molecule [14, 20]. H2O2 holds the –O—O– single weak bond that can easily break via. ionization, such as the Fenton reaction, to generate the most reactive hydroxyl radical (OH•–) [21]. OH•– possesses the highest potential 1-electron reducing capacity in biological systems and reacts with different macromolecules in the vicinity of their production site [3, 14, 20]. OH•– can react with molecules through electrophilic addition and/or hydrogen abstraction. Pyrimidine bases of DNA, C – H, and S – H bonds in biological systems are examples of the prime targets of OH• [13]. On the other hand, peroxynitrite (ONOO−) represents a powerful oxidant among the reactive nitrogen species (RNS) that arises from the rapid reaction of NO• with O2•− [3, 10, 14, 22].
While systemic ROS accumulate from the contributions of several body tissues and organs, skeletal muscles are suggested to be the principal source of ROS during exercise [4, 14]. Exercise type, intensity, and/or duration are key factors that determine the magnitude and the endogenous source of ROS production [4]. Prolonged and intense exercise results in an irritated intracellular environment such as elevated body temperature, reduced cellular pH, increased CO2 tension, and decreased O2 tension associated with increased cellular respiration, all of which contribute to the increased rate of ROS production [23]. Such changes to homeostasis are suggested to increase ROS production essentially through three major endogenous subcellular mechanisms: mitochondrial electron transport chain, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and xanthine oxidase [4, 10].
Mitochondria, under normal physiological conditions, produce moderate amounts of superoxide anions at complexes I and III of the electron transport chain to the matrix and intermembrane space [24]. It was originally proposed that about 2 – 5% of the total oxygen consumed by mitochondria gives rise to O2•− during mitochondrial metabolic reducing state (State 4) [25], and the ROS production is largely increased during the active respiring state (state 3) in proportion to fold change in oxygen consumption [26]. However, more recent findings have suggested that the mitochondrial contribution to the increased oxidative stress during exercise is minimal. For example, only 0.15% or less of the total oxygen consumption is reduced to O2•− under resting partial pressure of oxygen [24]. Furthermore, data from isolated mitochondria indicated a higher rate of O2•− production at basal state 4 compared to the respiring state 3 in skeletal and diaphragm muscles [22]. Mitochondria in vivo are, in most part, close to or in state 3, in which mitochondrial ROS generation is negligible [4, 22, 27]. In addition, confocal microscopy with specific fluorescent probes technique has shown that mitochondria do not contribute to the cytosolic O2•− accumulation neither at rest nor following muscle contractions [28].
NADPH oxidase Nox1, Nox2, and Nox4 isoforms are expressed in skeletal muscles where Nox2 and Nox4 are the most abundant homologues. Nox2 isoform is localized at the sarcolemma and transverse tubules of the skeletal muscles, where it primarily modulates the cytosolic superoxide during rest and muscle contractions [28]. Muscle fibers incubated with the NADPH oxidase inhibitor apomycin reduced the cytosolic fluorescence of 2-hydroxyethidium by ~10% and ~70% in the resting and contracting fibers, respectively, compared with vehicle-treated fibers [28]. Thus, for these data and others, NADPH oxidase has been suggested as the major source of superoxide generation in contracting skeletal muscles [29].
Xanthine oxidoreductase is an enzyme that exists in two isoforms; xanthine oxidase and xanthine dehydrogenase that both catalyze the oxidation of hypoxanthine to xanthine and xanthine to uric acid. Under normal conditions, xanthine dehydrogenase is the principal isoform that uses NAD+ as the oxidizing agent. On the other hand, during intensive exercise, xanthine dehydrogenase is converted to oxidase, which uses O2 as an electron acceptor to form O2•−. This conversion is dependent on the increased levels of intracellular calcium and oxidants during intensive exercise [30]. It has been suggested that activation of xanthine oxidase occurs during intensive exercise in a manner similar to ischemia-reperfusion [31]. There has been a controversy concerning the role of xanthine oxidase in contracting skeletal muscles. For example, maximal contraction force was attenuated, and oxidative tissue damage was increased during exhaustive muscle contractions in rats and mice. Both responses were partially or completely reversed by xanthine oxidase inhibition, suggesting a key role xanthine oxidase plays in exercise-induced oxidative stress [31, 32]. In contrast, some have suggested a limited role for xanthine oxidase in exercise-induced oxidative stress since human skeletal muscles generally express low amounts of xanthine oxidase and dehydrogenase [10, 29]. Nonetheless, xanthine oxidase can be increased in skeletal muscles via microvascular endothelium and leukocytes infiltration, by which it may compensate for the low expression of the enzyme in the muscle fibers [33].
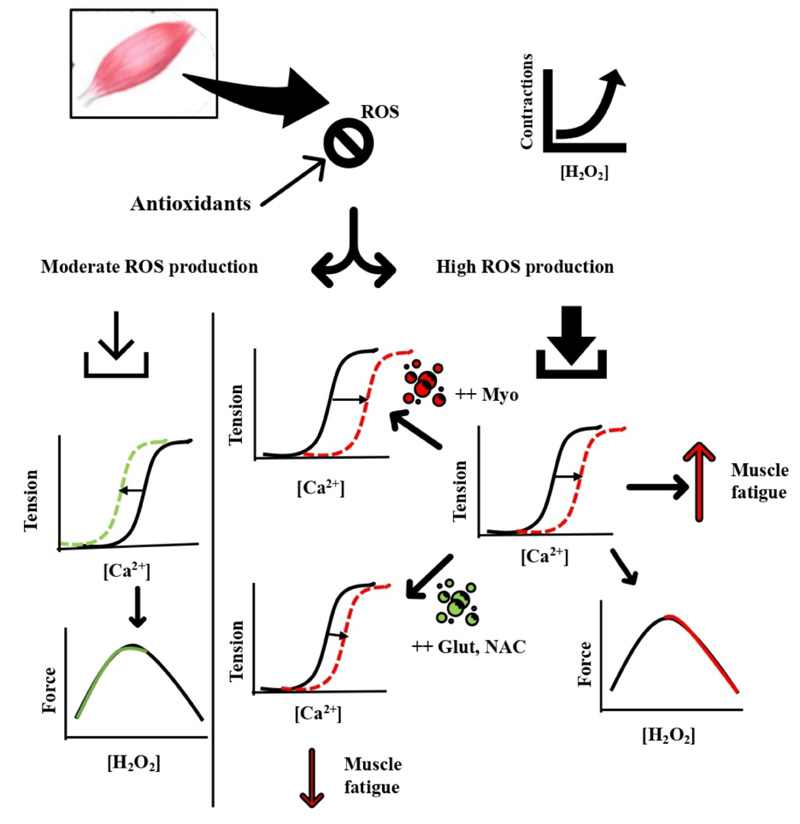
2.2. The Damaging Effects of Oxidative Stress during Exercise
ROS production under normal physiological conditions is counteracted and controlled by the cellular antioxidant defense system. ROS at basal levels is implicated in mitogenic proliferation, cell signalling, and regulation [34]. Fig. (2) summarizes the damaging effect of oxidative stress induced by muscle contractions during different modalities of exercise. ROS production during moderate muscle contractions is well counteracted by antioxidants and contributes to increasing the myofibrillar Ca2+ sensitivity and force production [35, 36]. During fatiguing muscle contractions, ROS production rises to a level where cellular antioxidants become overwhelmed, and subsequent disruption of cellular functions and integrity is prompted [37] (Fig. 2). The increased oxidative stress during exercise has been implicated in impaired exercise performance and systemic functions. This was first illustrated by a model showing that isometric force during fatiguing exercise progressively declines with increasing levels of ROS in a dose-dependent manner [38]. The contribution of exercise-induced ROS production in muscle fatigue has been depicted from the findings related to antioxidant supplementations and their effects on delaying muscle fatigue [10, 14, 38]. For example, endurance athletes receiving the antioxidant compound N-acetylcysteine (NAC) improved their submaximal cycling time to exhaustion by ~26% compared with the placebo trial [39].
Moreover, respiratory muscle fatigue was reduced with NAC versus placebo infusion during breathing through the inspiratory resistive load, and time to fatigue increased from ~13 min with placebo to ~21 min with NAC infusion [40]. In addition, iron injections to accelerate ROS production increased muscular malondialdehyde, a sign of lipid peroxidation, which resulted in decreased endurance performance and muscular force generation along with muscle atrophy [41]. Others have shown that superoxide dismutase-deficient mice increased the levels of oxidative damage and impaired exercise performance without muscle atrophy [42]. These data suggest the increased ROS production is pivotal in inducing muscle fatigue since antioxidants supplementation ameliorated the reduced muscle performance that coincides with the increased oxidative stress.
The mechanisms underlying the ROS-induced muscle fatigue development are proposed to involve effects on the sarcoplasmic reticulum (SR) channels and myofibrillar components. Sulfhydryl groups of the SR ryanodine receptors are highly susceptible to H2O2 oxidation, which upon oxidation, activates Ca2+ release [43]. Others have shown that H2O2 primarily affects muscular contraction through changing myofibrillar Ca2+ sensitivity rather than SR Ca2+ release [44]. Brief exposure to H2O2 (100 – 300 μM for 4 min) transiently increased myofibrillar Ca2+ sensitivity and contractile force generation; when the H2O2 exposure time was prolonged to 8 min, both myofibrillar Ca2+ sensitivity and force production were reduced. All responses were not accompanied by changes in intracellular Ca2+ concentration [44]. The impact of H2O2 on contractile function and Ca2+ sensitivity has been found to be largely dependent on the presence or absence of myoglobin and glutathione [10, 45]. Using skinned fibers, H2O2 had a minimal effect when myoglobin was absent. In fibers rich in myoglobin, H2O2 induced a remarkable reduction in Ca2+ sensitivity and force production, possibly through the Fenton reaction with myoglobin Fe2+ to produce the highly reactive hydroxyl radical [46]. Glutathione, on the other hand, when added to skinned muscle fibers, reverses the decline in Ca2+ sensitivity and forces production via S-glutathionylation of a particular cysteine residue (most likely Cys133) on the troponin I isoform. This effect was also observed in exercising muscles in humans [47].
ROS-induced cell damage and apoptosis are the results of ROS attacking all types of cellular macromolecules. Polyunsaturated lipids of phospholipids bilayer of every cellular membrane and other cellular components are highly susceptible to oxidative damage that may cause disruption to membrane integrity and permeability [2, 48, 49]. ROS can readily attack several amino acid residues in proteins such as tyrosine, methionine, tryptophan, histidine, and sulfhydryl residues, most often resulting in irreversible protein degradation and aggregation [48]. Importantly, nucleic acids of DNA and RNA are highly susceptible to oxidative damage. ROS attacks have been suggested to induce DNA lesions at rates of 10,000 and 100,000 per day per cell in humans and rats, respectively [50]. Accumulation of DNA lesions as a result of a higher rate of oxidative DNA damage and a lower rate of enzymatic excision of DNA lesions has been implicated in several conditions such as mutagenesis, carcinogenesis, and aging [50, 51].
2.3. Mechanisms of Oxidative DNA Damage
Both OH• and ONOO− are strong oxidizing agents that cause oxidative DNA damage by addition, H-abstraction, and/or electron transfer to and from the nitrogenous bases and sugar moiety of DNA. OH• is the most reactive radical, with an estimated 70% of this radical reacting with the nitrogenous bases and 30% with the deoxyribose moiety [52]. All four DNA nitrogenous bases are susceptible to OH• attack, typically through addition to C—C and C—N double bonds and to a lower extent through H-abstraction from the 2`-deoxyribose moiety. Those reactions with DNA nitrogenous bases give rise to a wide multiplicity of adduct base radicals [53]. Pyrimidine bases, thymine, and cytosine are susceptible to OH• addition at C5- and C6- positions, respectively, by which OH-adduct radicals are formed. In addition, thymine undergoes H-abstraction from the methyl group, which forms allyl radicals. The C5-OH adduct possesses reducing and C6-OH adduct oxidizing redox properties [54]. C5-adduct radicals can readily undergo oxidation followed by OH− addition, or water addition followed by deprotonation giving rise to thymine glycol and cytosine glycol formation. Thymine glycol and cytosine glycol can also be formed by the addition of oxygen to C5-OH adduct radicals to yield 5-hydroxy-6-peroxyl radical that may undergo superoxide elimination followed by OH− addition. C5-OH and C6-OH adduct radicals can be reduced and protonated in the absence of oxygen to 5-hydroxy-6- and 6-hydroxy-5-hydropyrimidines. Further, uracil derivatives might be yielded from additional deamination and dehydration particularly to cytosine products, such as cytosine glycol. Uracil derivatives may also be formed through oxidation of the allyl radicals of thymine [51, 55].
Purine nucleic acids are susceptible to OH• addition at C4, C5, and C8 positions yielding OH-adduct oxidizing and reducing radicals. C4- and C5-OH adduct radicals undergo dehydration and subsequently convert to oxidizing purine radicals, which may be reduced to reconstitute purines. C8-OH adduct radicals give rise to yield formamidopyrimidines and 8-hydroxypurines upon one-electron reduction and oxidation, respectively [51]. 8-hydroxypurines can be yielded in the absence or presence of oxygen but more favourably in the presence of oxygen. 8-hydroxydeoxyguanosine (8-OHdG) is a significant product of C8-OH adduct radical oxidation. 8-OHdG is the predominant oxidatively-modified DNA base among more than 100 other modifications and the most widely used biomarker in the measurement of DNA oxidation levels. The popular use of 8-OHdG in the detection of oxidative DNA damage is in part due to the simplicity of methodology. For instance, several mechanisms contribute to excising 8-OHdG from DNA strands, preventing its incorporation into cellular DNA, and subsequently excreted to blood circulation and urine [56, 57]. The excreted 8-OHdG residues are electrochemically active and can be easily detected using high-performance liquid chromatography with electrochemical detection method (HPLC-EC) in the serum and urine [58]. In addition, 8-OHdG within the DNA strands can mispair with adenine, which subsequently causes GC to AT transversion to form the most common mutation site of human cancers [56]. Accordingly, 8-OHdG has been found to be highly correlated with the development of various types of cancer and degenerative diseases [51, 55, 58].
All five DNA deoxyribose centered carbons are prone to H-abstraction by OH• according to their steric accessibility to OH• in the following order of reactivity: 5´ H > 4´ H> 3´ H ≈ 2´ H ≈ 1´ H. H-abstraction from the deoxyribose moiety results in DNA lesions with numerous sugar termini such as 3´-phosphate, 3´-phosphoglycate, 5´-phosphate, and 5´-aldehyde [59]. C5´ represents the largest DNA cleavage site, which undergoes an intramolecular cyclization through addition to the purine ring at C8-position of the same nucleoside followed by oxidation [51, 55, 59].
3. EXERCISE-INDUCED OXIDATIVE DNA DAMAGE
Factors known to increase oxidative stress have been shown to augment oxidative DNA damage. For example, smokers, by means of increased ROS production, exhibit greater DNA damage biomarkers than non-smokers [60]. Moreover, factors related to decreased metabolic rate such as being a female, obese, aging, and on calorie restriction have been associated with lower levels of oxidative stress and oxidative DNA damage [61-64]. Exercise as a potent stimulant of ROS production can acutely cause oxidative DNA damage. The magnitude of this damage might be dependent on exercise components such as type, duration, intensity, and volume.
3.1. Oxidative DNA Damage Responses to Resistance Exercise
Several muscular changes and events that occur during resistance exercise can potentially increase ROS production. Particularly, the active muscle during resistance exercise involves brief and frequent ischemia-reperfusion periods, membrane disruption associated with increased Ca2+ influx, and elevated inflammatory mediators. All of those events have been linked with increased levels of ROS [2, 10]. Resistance exercise effects on the oxidative damage of lipids and proteins were extensively studied [7, 65], while less attention has been given regarding oxidative DNA damage.
High-intensity whole-body resistance exercise protocols have been consistently shown to elicit oxidative DNA damage in a variety of populations. A single session of high-intensity whole-body resistance exercise (7 sets of 4 exercises at 80 – 90% 1RM) increased urine 8-OHdG in resistance-trained subjects immediately and at 24 h postexercise relative to pre-exercise level [66, 67]. Similarly, 4 sets of 4 whole-body multi-joint resistance exercises increased urine 8-OHdG immediately, 3h, and 24 h postexercise in untrained men, and at 3 h and 24 h post-exercise in trained men. Oxidative urine 8-OHdG was higher in the untrained group [68]. Moreover, performing 4 rounds of 4 power exercises at 80 – 90% 1RM using cluster or traditional set configurations immediately increased the serum 8-OHdG in resistance-trained volleyball players [69]. It is evident that whole-body resistance exercise can elicit oxidative DNA damage in trained and untrained subjects, but to higher levels in the untrained.
Regional resistance exercise protocols, on the other hand, have demonstrated inconsistent responses of oxidative DNA damage indices. Sixty maximal eccentric contractions of the elbow flexors did not change serum 8-OHdG, despite the increased levels of protein carbonyls, total oxidant status, oxidative stress index, and muscle damage [70]. Likewise, squatting at 70% 1RM for about 90 repetitions performed within 30 min increased plasma protein carbonyls at 6 and 24 h post-exercise, but not serum 8-OHdG [71]. In contrast, 200 eccentric contractions at 60% of maximal isometric contraction of rectus femoris induced muscle soreness associated with 24 h postexercise increase in 8-OHdG content of skeletal muscle [72]. The above controversial findings might be related to the total volume of exercise and the medium where 8-OHdG was measured. During regional resistance exercise, it seems that a higher volume, i.e., sets and repetitions, which implies a longer duration under stress, may induce a significant elevation of oxidative DNA damage. Oxidative DNA damage biomarkers were not changed when performing 60 – 130 repetitions [70, 71], whereas the 8-OHdG level was only increased following 200 contractions [72]. In addition, the site of 8-OHdG detection might have influenced the findings. The 8-OHdG increase was observed when measured in the muscle tissue [72], while it was not detected when measured in serum following resistance exercise [70, 71]. Serum level of 8-OHdG represents the systemic DNA oxidative damage, which requires bigger muscle mass involved in the exercise; hence, resistance exercise utilizing small muscle mass might not be able to elevate the serum 8-OHdG level. Nonetheless, the muscle mass and training volume in resistance exercise remain critical factors to induce oxidative DNA damage since only serum oxidative DNA damage biomarkers were absent while increased protein and lipid oxidative damage biomarkers were evident following regional resistance exercise protocols.
The variation in exercise-induced oxidative DNA damage during regional resistance exercise might not be related to the training status of the participants. 8-OHdG was not changed in trained [71] and in nontrained subjects [70], while it was increased in recreationally active subjects [72]. Also, oxidative DNA damage does not seem to be related to the muscle inflammation associated with eccentric exercise because 8-OHdG was elevated prior to symptoms of delayed-onset of muscle soreness and was not changed despite increased muscle damage biomarkers [70, 72]. Altogether, this highlights the role of training volume and time under oxidative stress during resistance exercise in the induction of oxidative DNA damage.
The increase in 8-OHdG ranged from < 1 to about 2 ng/ml that peaked at 24 h postexercise in all the utilized whole-body resistance exercise protocols [66-69], and about 0.09 pmol/
Regular resistance exercise training has been shown to confer protection against exercise-induced oxidative DNA damage. While basal urinary 8-OHdG was not different between groups, trained young men have demonstrated lower levels of 8-OHdG at the post and 24 h post-whole-body high-intensity resistance exercise than untrained men [68]. In older adults (60 – 75 years), a whole-body resistance exercise training 14 – 16 weeks reduced basal urinary 8-OHdG without altering the redox state, antioxidant enzymes, or mitochondrial DNA deletion [73, 74]. In addition, DNA repair enzymes and DNA strand breaks were lower in trained compared to untrained subjects [75]. The reduced 8-OHdG might be attributed to the enhanced antioxidant capacity that was shown to increase after resistance exercise training [76]. Moreover, antioxidants supplementation reduced oxidative damage to lymphocyte DNA [77]. However, this might not be similarly working in all individuals since the antioxidants capacity was not changed in older- and/or middle-aged groups following resistance exercise training [73]. There might be other protective mechanisms against oxidative DNA damage rather than the antioxidant defense system that develop during resistance exercise training. One example might be an increased DNA resistance to oxidative damage through increased non-specific DNA binding protein Dps. Dps is a 19-kD protein that was first found in bacteria to bind on a non-specific DNA sequence to form a stable and DNase resistant complex [78]. Dps was directly found to reduce DNA single-strand breaks in vivo and DNA lesions after H2O2 treatment [79]. Whether Dps plays a protective effect against oxidative DNA damage at baseline and during exercise in skeletal muscles is still unexplored (Fig. 3).
3.2. DNA Oxidative-damage Responses to Aerobic Exercise
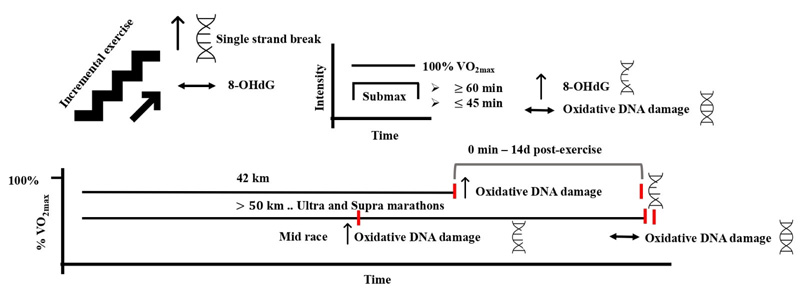
Aerobic exercise is defined as any physical activity that utilizes large muscle groups for prolonged periods of time. It is dynamic and rhythmic in nature and ranges from moderate to vigorous intensity [1, 80]. Adaptations to the cardiovascular and skeletal muscles in response to aerobic exercise have been widely reported [80-82]. Specifically, several studies have looked at the effects of aerobic type of exercise on the level of oxidative DNA damage with controversial results. This controversy is broadly attributed to the methods used to detect the levels of DNA damage, exercise structure, and the environmental conditions where the exercise was performed. Incremental running to exhaustion that was ~ 17 – 21 min in duration induced DNA damage in peripheral white blood cells using the single-cell gel electrophoresis assay in trained, untrained, and recreationally active subjects 24 h post-exercise [83, 84]. In another study, incremental exercise to exhaustion resulted in single-strand breaks of 10% of lymphocytes DNA immediately after exercise, but not at 24 or 48 h post-exercise [85]. DNA damage following this type of exercise seems to peak at 24 h and decrease to a pre-exercise level at 72 h postexercise [86]. On the other hand, oxidative DNA damage measured by the level of urinary 8-OHdG was not aggravated by incremental running and cycling to exhaustion in trained and untrained subjects at any time after exercise [87, 88]. In these studies, the level of DNA damage returned to pre-exercise level after 24 – 72 h postexercise, which may suggest this DNA damage is transient and repairable following an exhaustive aerobic exercise.
Sub-maximal endurance exercise appears to induce DNA damage when it is performed for extended durations, i.e., greater than 60 min. This was evident during cycling at 70% VO2max for 60 – 90 min, where urinary 8-OHdG was increased following exercise in moderately trained participants [89, 90]. Shorter durations of exercise (30 – 45 min) at 50 – 85% VO2max failed to induce changes in any of the oxidative DNA damage biomarkers detected by several methods, including micronucleus, single-cell gel electrophoresis (comet), and HPLC-EC assays [71, 86, 91-93]. The moderate type of aerobic exercise generally seems incapable of inducing oxidative DNA damage under normal conditions and within 60 min of duration. This might be attributed to the findings that showed no increased oxidative stress following moderate aerobic exercise [94].
On the other hand, ultra-endurance exercise protocols have shown remarkable increases in oxidative DNA damage. A marathon race resulted in increased DNA single-strand breaks, the amount of oxidized purines, and pyrimidines using formamidopyrimidine glycolase (FPG)- and endonuclease III-sensitive sites, respectively, and the urinary 8-OHdG immediately and until 14 days after the race [95]. During a 50 km ultramarathon, DNA damage in leukocytes assessed by comet assay was increased in the mid-race and returned to baseline at 2 h postexercise [96]. Moreover, the supra-marathon (130 km over 2 days and 328 km over 4 days) increased urinary 8-OHdG level after the first day of the race, which returned to baseline by the end of the race days [97, 98]. These findings suggest that ultra-endurance exercises do not result in prolonged DNA damage, likely through enhanced DNA repair and antioxidant defense systems. That was shown during the ironman triathlon, where the plasma antioxidant capacity, assessed by the ferric reducing ability of plasma and oxygen radical absorbance capacity assays, peaked immediately post-exercise and declined thereafter [99]. The propagated DNA damage observed after a marathon race compared with the other ultra-and supra- distances could also be explained by the intensity of exercise. The median finish time (~ 3 h) of the marathon race necessitates a higher intensity performance compared with longer durations (7 h – 4 days) of ultramarathon and supra-marathon races that require the athletes to perform at lower intensities [95-98]. The higher intensity during long durations of exercise might have caused an overwhelming increase in oxidative stress, in which the antioxidant and repair systems were unable to cope with (Fig. 4). Alternatively, the oxidative DNA damage was assessed in white blood cells during the ultramarathon [96] that are not the main site of oxidative stress during exercise. Previous work could not draw a relationship between oxidative DNA damage in muscle cells and mononuclear cells. Hence, the DNA damage observed in white blood cells does not represent the damage in skeletal muscles, which could partly explain the difference between oxidative DNA damage during the marathon and the other ultra-and supra- endurance distances.

Extreme physiological and environmental conditions appear to augment the exercise-induced oxidative DNA damage following exercise protocols that were previously shown of modest or negligible effects to increase DNA damage. For example, incremental ergometer cycling to exhaustion at high-altitude hypoxia (4559 m for 3 days) resulted in increased leukocytes DNA strand breaks, urinary 8-OHdG, and endonuclease III-sensitive sites compared to no changes at normal conditions. The augmented DNA damage was possibly due to the increased leakage of mitochondrial ROS and/or hypoxia-induced inflammation under hypoxic conditions [100]. Furthermore, running to exhaustion at 80% VO2max (~ 28 – 40 min) induced greater lymphocytes DNA strand breaks during dehydrated compared to euhydrated state, probably due to dehydration-induced higher thermal stress [101]. Other pathological and environmental conditions such as diabetes and heat stress were associated with increased levels of oxidative DNA damage [102, 103], and hence they have a high potential to increase the exercise-induced DNA damage. Nonetheless, the effects of exercise under heat stress and pathological states on DNA damage are lacking and need to be elucidated.
Chronic aerobic exercise, like resistance exercise training, appears to augment adaptations to lower oxidative stress and the associated oxidative DNA damage. Physical fitness level was suggested to play a critical role in determining the changes in urinary 8-OHdG during ultra-endurance exercise [98]. DNA strand breaks following an ironman triathlon race were less prominent with the higher training status of the participants [99]. In addition, DNA strand breaks indicated by the comet assay were lower or completely prevented in trained compared with untrained subjects [83]. Moreover, setting the body at a negative energy balance through 20% exercise over a year substantially reduced oxidative DNA and RNA damage in white blood cells by 46% and 52%, respectively, in men and women aged 50 – 60 years [92]. Similar to resistance exercise, the mechanisms underlying the effects of aerobic exercise in reducing basal and exercise-induced DNA damage are still not fully explored. Possible mechanisms might be an enhanced activity of DNA repairing enzymes and antioxidant defense system [104, 105].
4. PERSPECTIVE AND FUTURE DIRECTIONS
Despite the fact that exercise may robustly increase oxidative DNA damage, there is no evidence that this damage will be permanently incorporated into DNA strands and alter the human genome. The increased DNA damage with exercise was repaired within hours to days, and furthermore, in some cases by the end of the exercise. Impaired DNA repair was not evident in any of the reviewed protocols, including high-intensity whole-body resistance exercises and ultra-endurance events. Conversely, the current evidence may indirectly indicate a protective role of exercise against mutagenesis. For example, data has shown muscle impairment during aging was associated with genome mutation [106], while exercise has been widely reported as a countermeasure against muscle impairment associated with ageing [107]. In addition, physical activity has been shown as a primary intervention to prevent cancer development and the associated mutagenesis [108, 109]. Nevertheless, analysing the human genome of skeletal muscles and other sites that are vulnerable to oxidative DNA damage during exercise in physically active and non-active subjects is highly warranted.
Most of the work, if not all, was conducted on exercise-induced DNA damage in skeletal muscles or in the systemic circulation. Other active organs during exercise, most importantly the heart, have not received sufficient attention examining the level and type of oxidative DNA damage that may occur during exercise. The susceptibility or resistance of these organs to damage and whether such damage, if any, results in permanent changes to the DNA structure are areas of research that warrant more studies. Moreover, the responses of oxidative DNA damage to exercise in different pathologies such as diabetes are still missing. Pathological conditions might exacerbate exercise-induced oxidative stress and the resultant DNA damage. Since redox state and antioxidant capacity were not changed in some populations [73, 74], more attention should be given to the factors that protect DNA from oxidative damage, such as the DNA non-specific binding protein Dps.
CONCLUSION
Both aerobic and resistance exercise types are potent stimuli that increase oxidative DNA damage. However, resistance exercise appears to prompt greater oxidative DNA damage when performed at higher load, higher total volume of sets and repetitions, and involve larger muscle groups. Aerobic exercise seems to induce oxidative DNA damage during higher intensities and longer durations (i.e., longer > 60 min). There is currently no evidence suggesting that the acute exercise-induced oxidative DNA damage can be incorporated into permanent alterations of the human genome. Rather, regular exercise training appears to induce adaptations that protect against oxidative DNA damage and possibly mutagenesis.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by the Scientific Research and Graduate Studies deanship at Yarmouk University under Grant No. 4424-4-11.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The author is grateful to Iris Bessiouni and Mohammad Ihsan for proofreading the article.

